
- 论文原文:HiPiler: Visual Exploration of Large Genome Interaction Matrices with Interactive Small Multiples
- 作者:Fritz Lekschas, Benjamin Bach, Peter Kerpedjiev, Nils Gehlenborg, and Hanspeter Pfister
- 发表刊物/会议:2017 InfoVis
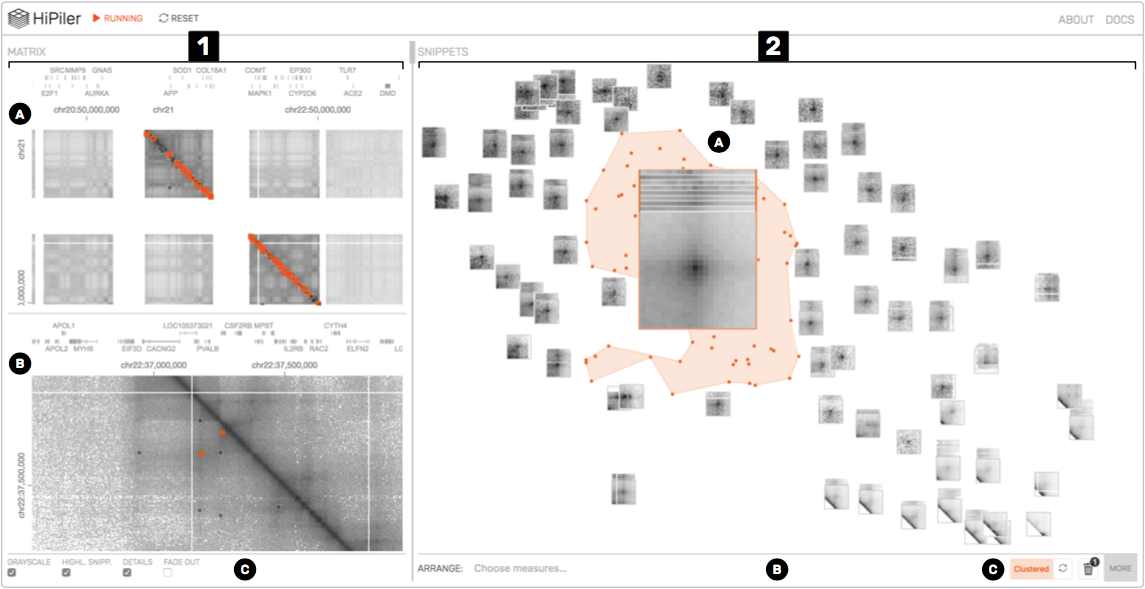
图 1:系统界面。矩阵视图(1)包含概述(1A)和详细(1B)矩阵。 片段视图(2)将矩阵的区域呈现为交互式的 small multiples。 在这个例子中,片段以 t-SNE(2C)排列,一堆片段会以其平均模式进行显示(2A)。 查看操作菜单位于底部(1C 和 2B)。
个人评述
- 结合了 small multiple 和 overview+detail 的方法来探索大矩阵,对大规模数据可视化很有借鉴意义。
- 文章整体领域性特别强,就很注重讲故事的能力,并且整篇文章的讲故事能力也很强。
启发:
这对大规模图可视化其实有一个很好的借鉴意义,但相比于文中的有序矩阵,图的无序性会让其在拓扑空间的上下文很难展示在平面上。
overview->detail->small multiple 的方法,是大规模数据可视化的一个重要模式。但在图可视化中,这几个部分都还很具有挑战性。
结合领域知识,使整片文章的 motivation 非常充足,或许也给我们一个思路,在结合领域的基础上进行科研,但又要跳出领域知识,比如这篇文章并没有把自己局限在生物领域,在其他领域,只要满足其矩阵有序的基本假设,都可以应用这个方法。
介绍
生物学家对基因序列是如何折叠的研究非常感兴趣,可以帮助理解细胞的功能、疾病的产生等。
基因组之间存在相互作用(_interacting_),两两之间相互作用则会形成一个基因组相互作用矩阵(_genome interaction matrix_),最多能达到三百万 × 三百万。里面会包含一些重复出现的图案/模式(称为ROI, _regions of interest_)。
- 挑战:
- 算法识别的结果不是很可信,并且没有ground-truth去评估这些算法是否准确。
- 成千上万的小ROI的探索和比较,还没有工具能够支持。
- 贡献:
- 对算法检测到的团进行可视化评估
- 大规模图案集合的分类、聚合、异常点检测
- 在多个矩阵之间比较ROI(比如,在不同数据集,实验条件,抽取算法等等),_下文中也会称之为 Snippet_
- 对其他的基因属性和图案之间的相关性进行分析
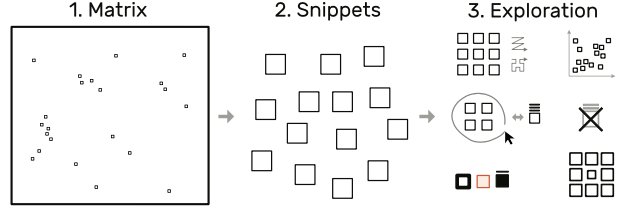
图 2:snippets方法,将大矩阵(1)分解成小片段(2),并使用不同的布局,排列和样式探索这些片段(3),同时保持全局上下文。 矩阵内的小方块表示片段位置。
背景
Hi-C 矩阵分析:HI-C是一种捕获基因序列片段之间相互交联的方法。基因序列片段如果在空间上如果比较接近的话,会相互交联,如下图。每次检查数百万个不同细胞,会产生基因序列片段之间的平均接触概率矩阵。矩阵非常稀疏,并呈现这样的规律:空间邻近的基因序列片段更容易相互交联。
矩阵元素的颜色深浅编码了基因片段之间的接触概率。
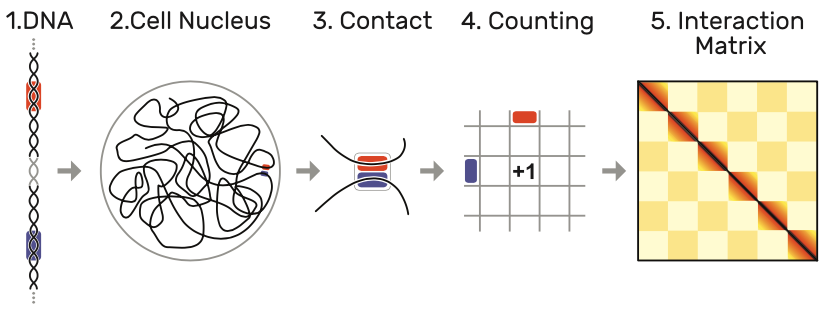
图 3:Hi-C 方法:由于 DNA(1)在细胞核(2)中并非随意进行组织的,某些序列会进行紧密交联(3)。在经过数亿个细胞计数之后(4),会得到一个多达 300 万个单元的相互交联矩阵(5),深色表示交联次数更多
专家采访:主要围绕了以下三方面展开,
基因折叠相关研究的长期目标
更好地理解基因组对基因调控等过程的作用。
工作的流程和策略
当前的挑战
- 不同 ROI 相距甚远,普通工具容易导致上下文的丢失。
- 很多图案之间差异细微
- 数据噪音的存在,并非所有的图案都跟生物特征有关(有些仅仅是因为空间限制引发了交联)
常见的 Hi-C 矩阵图案:
下图显示了一些常见的图案:
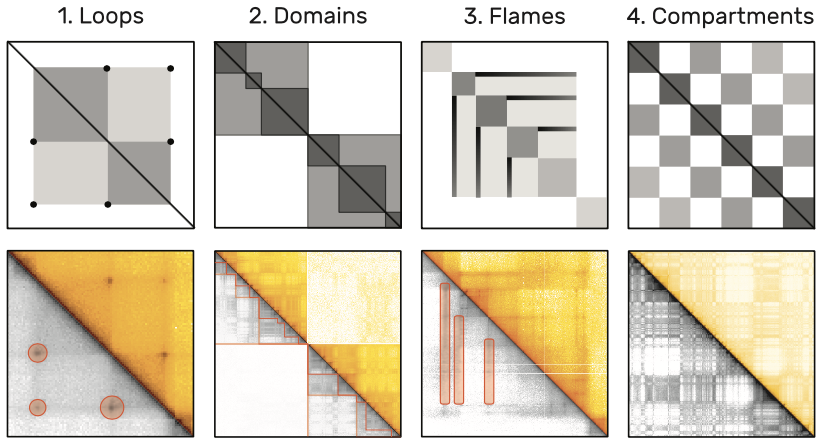
图 4:放大尺寸后的交联矩阵中的频繁图案的示例。上方的矩阵是下方矩阵的一个示意。交联矩阵是对称的,所以下面的三角用灰度来显示,以突出那些用橙色标记的图案。
分析任务:
T1. 搜索已知的图案
T2. 发现新的图案
T3. 对某个图案实例进行研究
T4. 比较单个图案类型中包含的多个实例
T5. 将图案和其他生物特征进行关联
T6. 比较矩阵中不同的ROI
相关工作
围绕了:
- 基因交联矩阵可视化
- 大矩阵可视化
- 分而治之的挖掘方法
HiPiler 的设计
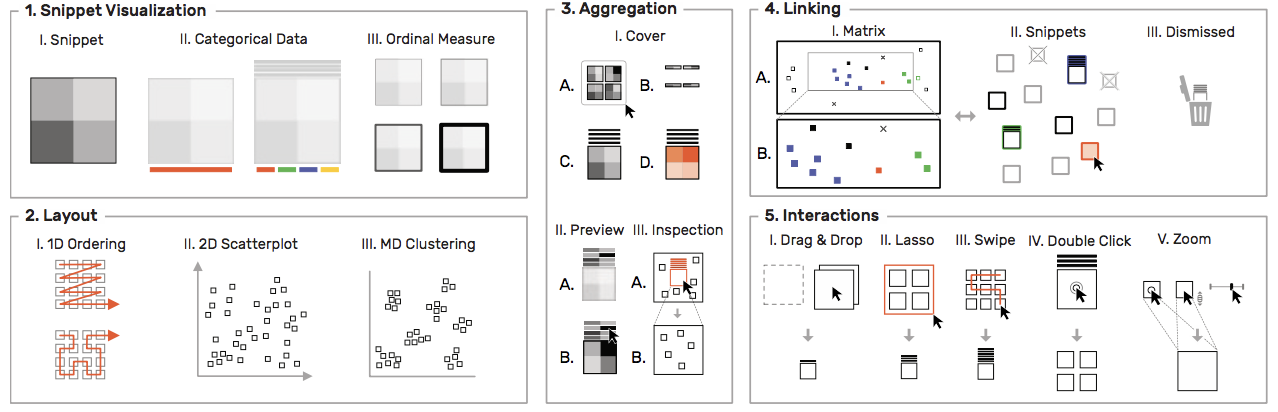
图 5:HiPiler 的可视化和交互的概览
关键目标:利用smal multiple的方法来对大矩阵的多个ROI进行探索。(如何提取ROI不是本文工作)
Hipler的设计过程是被以下几个问题所驱动的:
- 如何有意义的限制ROI的显示数量?
- 用什么交互来支持有效的排列?
- 如何链接这些ROI和整个大矩阵?
数据模型
这里假设了矩阵的行和列的排序是固定的,由基因序列给出。
设计
跟计算生物学家之间讨论,研究了不同的可视化设计,并最终决定使用矩阵来进行可视化。
片段隐喻(Snipptet Metaphor)
解决任务T1,T2,T3,T5。用来展示ROI的的矩阵数据,附加的一些可视化设计可以编码类别信息和有序数据:类别信息会被编码成颜色放在底下,有序数据则可以被编码成边框的颜色和粗细。

图 6:片段snippet被绘制为热力图(1),显示其ROI的矩阵数据。 分类属性可以用颜色标签可视化(2)。 与片段相关的有序数据可以通过边框的宽度和颜色(3)显示。 例如,较高的值被绘制为黑色粗边框。
片段布局(Snippet Layout )
解决任务T4。通过降维的方式,允许进行一维排序(1D),二维的布局(2D)以及多维的聚类(MD)。

图 7:片段snippet可以沿着各种不同的尺寸排列。 对于单个属性,片段在支持读取方向和希尔伯特曲线(1)的 1D 中布置。 选择两个属性创建散点图(2)。 对于两个以上的属性,HiPiler 应用了降维算法(3),如 t-SNE [33]。
聚合(Aggregation)
解决任务 T4,T6。利用了打桩(_piling_)来隐喻多个snippet的聚合。聚类后,每个片段每列取平均值划为一行的形式显示在上方,可以 hover 查看具体图案。集合封面可以显示集合的平均值或者方差。
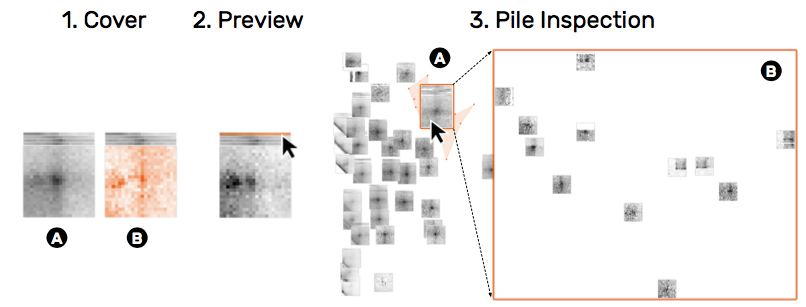
图 8:HiPiler 显示一堆片段的平均值(1A)或方差(1B)的封面矩阵。 此外,每个片段都显示为一维预览,显示片段列的水平聚合(2)。 将鼠标光标移到预览上可显示相关矩阵。 检查堆(3B)的时候暂时隐藏所有其他片段(3B)。
关联(Linking)
解决T5,T6。将细节视图中的snippet关联到大矩阵中,因为在基因序列上的邻近信息对分析很重要。通过颜色标记来标记snippet在矩阵中的位置,不在矩阵视窗中的那些snippet则会被淡出。
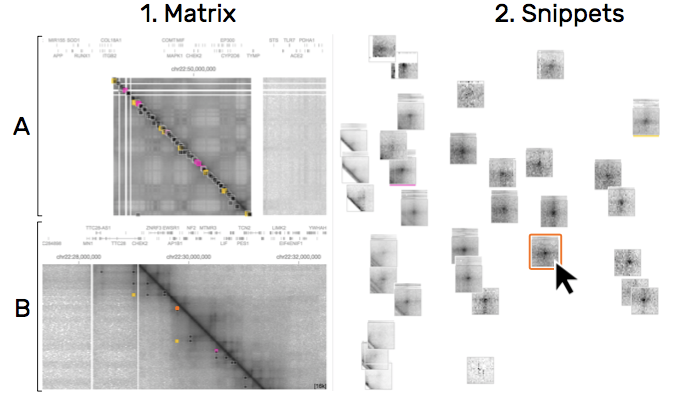
图 9:当在片段上进行悬停,该片段的位置会在交联矩阵(1A)和矩阵细节(1B)进行高亮显示。其他的颜色表示另外选择的矩阵。
交互技术
如图 5.5 所示,用户可以通过拖拽、lasso、刷选等方式来创建堆,双击以分散堆。缩放的时候,片段也会对窗口进行自适应。
评估
评估通过 USAGE SCENARIOS 和 USER EVALUATION 展开。
其中 USER EVALUATION 不同于之前的 USER STUDY,仅仅是让生物学家来使用这个系统,然后反馈意见。